 Login/Register
Login/Register 




Uncovering SARS-CoV-2 Specific Cells and Biomarkers
As we seek to better understand the mechanisms of SARS-CoV-2, it is important to have the right resources on hand to study the corresponding immune response generated by host cells. The first step involves the identification of participating immune cells, like T cells and B cells. This is followed by the thorough characterization and phenotyping of the cells, potentially leading to the discovery of new biomarkers that differentiate subpopulations, effectively opening the way to new treatments and therapies. In this blog post, we will look at new tools BioLegend provides to researchers performing this critical discovery work, including TotalSeq™ proteogenomics antibodies recently used by Su et al. in their Cell publication.
To stay current on our newest SARS-CoV-2 research tools, sign up for our eNewsletter and browse through our webpage for reagents.
TotalSeq™ Reveals Unique Subpopulations

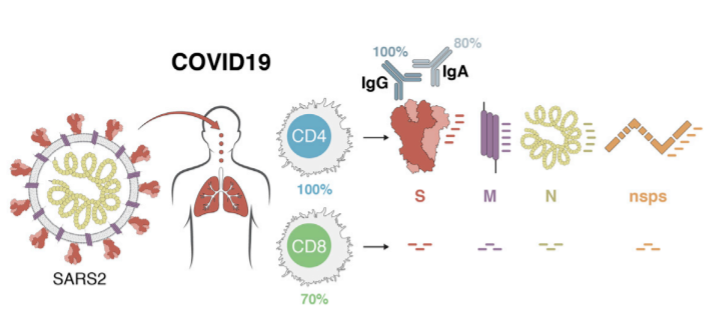
Su et al. began by gathering plasma and PBMC samples from healthy and COVID-19 afflicted patients. Patients were classified as mild, moderate, or severe based on the World Health Organization’s ordinal scale. While the group looked at hundreds of metabolites and secreted proteins, we’ll focus on key immune cell subpopulations they discovered using a panel of over 190 TotalSeq™ oligonucleotide-conjugated antibodies for single-cell multiomic analysis. Interestingly, they found several unique cell subtypes that began to appear in moderate cases, indicating a sharp distinction in the immune cell profile as patients progress from mild to moderate status. Throughout the article, T cell exhaustion is mentioned. T cell exhaustion describes a dysfunctional state where T cells progressively lose cytotoxic or anti-viral function. These cells become exhausted because they are continually exposed to antigens, such as in cancer or chronic viral infections. Phenotypically, these cells may alter their transcriptional and cytokine profile and upregulate exhaustion markers like PD-11. Knowing this, we can review some of the subpopulations identified below.
T cells: TCR profiling revealed two sub-populations of CD8+ T cells that correlated with disease severity: cytotoxic effector and memory-like phenotypes. The cytotoxic cells were found mainly in moderate/severe cases, while the memory-like phenotype was from mild patients. This indicates that cells may shift from a memory phenotype to focus on clonal expansion during the progression from a mild to moderate status. In addition, an exhausted and proliferative CD8+ T cell population emerged, as noted by expression of markers like LAG3, TIGIT, and CD279. Within CD4+ T cells, two subpopulations were revealed: one exhibited cytotoxic transcripts for PRF1 and GNLY, while the second featured exhausted and proliferative markers and a Th1 signature phenotype (similar to the CD8+ subpopulation of T cells noted above).
B cells: Patients with moderate/severe cases of COVID-19 possessed B cells that expressed more activation markers and had a larger expansion of antibody-secreting cells compared to healthy/mild cases.
Monocytes: Moderate patients displayed lower levels of non-classical (CD14lowCD16high) monocytes, but an increase in S100high HLA-DRlow monocytes. Su et al. theorize this latter group of monocytes may exhibit dysfunctional tendencies and contribute to a proinflammatory environment.
Natural Killer (NK) cells: NK cells in moderate patients become more cytotoxic-like, exhibiting a decrease in CD56 and CD127 transcripts, but an increase in PRF1, GZMB, and CD69 transcripts.
The shift in disease state from mild to moderate coincides with a rise in these rare cell populations and changes in the metabolic and cytokine profile. Taken together, these results indicate that the moderate stage may serve as the best potential time for therapeutic intervention.
Pinpointing SARS-CoV-2 Antigen Specific T Cells
T cells are a key component of the adaptive immune response against SARS-CoV-2. As new therapeutics and vaccine options are developed, such as the latest ones by Pfizer/BioNTech and Moderna, it is paramount to determine which portion of the virus T cells triggers the most efficient response. Our Flex-T™ MHC tetramer technology allows researchers to pinpoint antigen-specific T cells. Provided in monomer form and preloaded with SARS-CoV-2 spike, membrane, or nucleoprotein peptides, these reagents can be assembled into tetramers to stain reacting T cells. A short list of our options can be found below, but dozens more will be released in the coming months. Check our webpage often for updates.
| Flex-T™ Peptide | SARS-CoV-2 Component |
|---|---|
|
TLACFVLAAV |
Membrane protein (61-70) |
|
GMSRIGMEV |
Nucleoprotein (316-324) |
|
LLLDRLNQL |
Nucleoprotein (222-230) |
|
SIIAYTMSL |
Spike protein (691-699) |
|
ALNTLVKQL |
Spike protein (958-966) |
|
VLNDILSRL |
Spike protein (976-984) |
|
LITGRLQSL |
Spike protein (996-1004) |
|
VVFLHVTYV |
Spike protein (1042-1050) |
|
RLNEVAKNL |
Spike protein (1185-1193) |
|
NLNESLIDL |
Spike protein (1192-1200) |
|
FIAGLIAIV |
Spike protein (1220-1228) |
Additional Reagents for SARS-CoV-2 Research
In addition to the unique offerings above, we provide several immunoassays for your SARS-CoV-2 workflow, including: MojoSort™ for magnetic bead-based cell isolation; recombinant proteins, cytokines, and growth factors for bioassays; and LEGENDScreen™, which allows for the screening of hundreds of cell surface markers via flow cytometry. Although the methodology differs between the immunoassays, both LEGENDScreen™ and TotalSeq™-C Human Universal Cocktail, V1.0 have the ability to detect a variety of surface proteins. The latter reagent was utilized in the paper by Su et al. and is available as a ready-to-use cocktail with 130 markers and 7 isotype control antibodies.
In order for research to progress, a versatile set of tools must be made available to researchers, clinicians, and scientists. BioLegend will continue to develop these novel reagents so that we can fully understand each cell’s role in SARS-CoV-2 immunity.







Follow Us