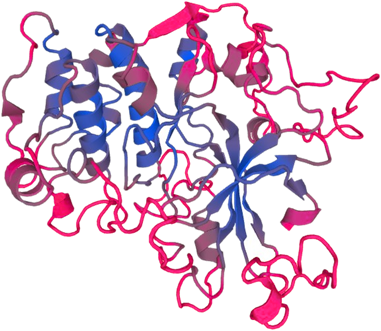
PTEN-induced putative kinase 1 (PINK1) is a serine/threonine protein kinase that is commonly mutated in familial forms of PD known as
autosomal-recessive juvenile Parkinsonism (AR-JP). PINK1 contains an N-terminal mitochondrial localization signal which allows its associated with the outer mitochondrial membrane where it interacts with its binding partners. PINK1 is responsible for surveying the mitochondrial health status by detecting and targeting damaged mitochondria for degradation.
Under healthy conditions where mitochondrial membrane potential is maintained, PINK1 is imported into the inner mitochondrial membrane (IMM) through interactions with mitochondrial translocase complexes, TOM and TIM. At the IMM, PINK1 is sequentially cleaved by MPP and PARL to remove its mitochondrial localization signal, and expose a region of PINK1 that signals it's targeting and degradation by the proteasome. This constitutive import and proteolysis of PINK1 ensures the maintenance of healthy mitochondria to meet the cellular energy demands. In damaged mitochondria, the loss of transmembrane potential leads to failure in transporting PINK1 to the IMM, and its accumulation at the outer mitochondrial membrane (OMM) where it remains bound to the TOM complex. Localization of PINK1 to OMM spares it from cleavage and subsequent proteasomal degradation. Instead, PINK1 undergoes autophosphorylation, and induces Parkin ligase activity and recruitment to the mitochondria. These proteins then initiate a cascade that leads to the degradation of damaged mitochondria through mitophagy. PINK1/Parkin-mediated mitophagy ensures the removal of toxic mitochondrial products and prevents neuronal loss. In PD patients, mutations in the PINK1 protein lead to accumulation of misfolded proteins in the mitochondria as well as fail to protect against stress-induced mitochondrial dysfunction.
 Login / Register
Login / Register 










 Mutations in the
Mutations in the 

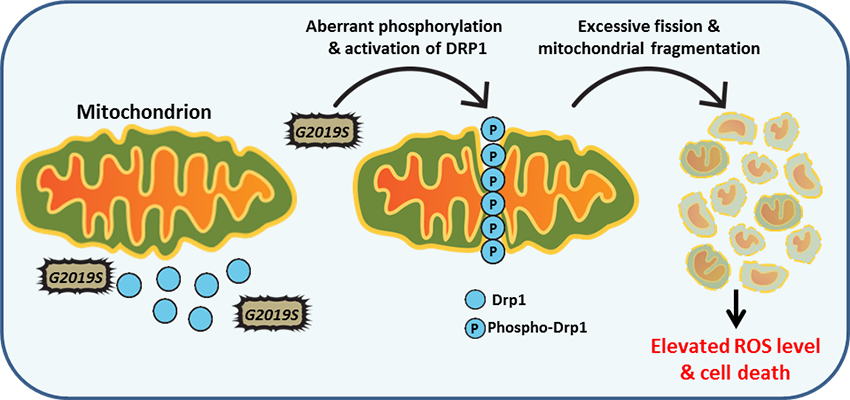
 Since its discovery in 2004, mutations in the
Since its discovery in 2004, mutations in the 
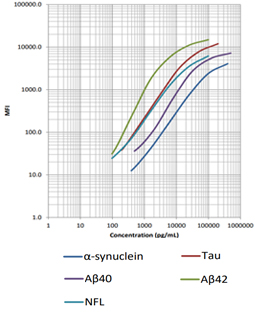
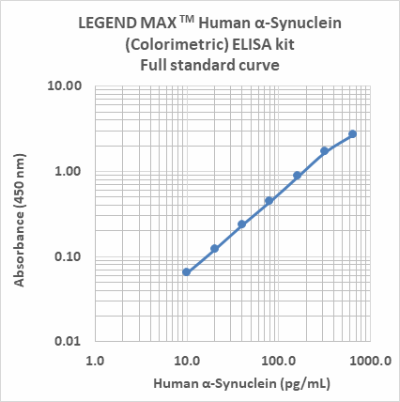





Follow Us